-
Characterization of transient radicals in the reduction product of the -P=C=C=P- system: EPR and theoretical studies
H. Sidorenkova, M. Chentit, S. Choua, M. Geoffroy and Y. Ellinger
Physical Chemistry Chemical Physics, 4 (20) (2002), p4931-4936


DOI:10.1039/b205848k | unige:3608 | Abstract | Article HTML | Article PDF
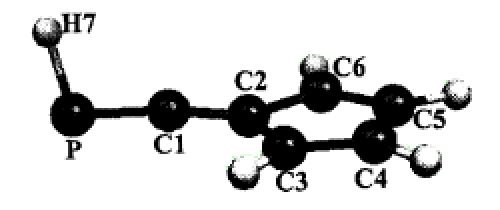
The EPR spectrum obtained at room temperature after electrochemical or chemical reduction of a solution of ArâP=C=C=PâAr in THF exhibits hyperfine interaction (165 MHz) with two equivalent 31P nuclei. Additional couplings with two equivalent 13C are observed with ArâP=13C=13C=PâAr. The 31P anisotropic coupling constants could be obtained from spectra recorded at low temperature. They indicate that the unpaired electron is mainly localized (78%) on the two phosphorus atoms. Quantum chemical calculations (DFT and ab initioSCI) were performed on the various isomers of the two radical anions: [HâP=C=C=PâH]â¢â and [HâP=CHâCH=PâH]â¢â. Although the optimized geometries of these two species are clearly different, neither of them leads to13C/31P hyperfine tensors in conflict with the experimental results. The absence of any 1H splitting on the EPR spectrum together with the quasi-reversibility of the reduction wave make the identification of [ArâP=C=C=PâAr]â¢â more probable.Â